Мукополисахаридоз – наследственное заболевание соединительной ткани, связанное с нарушением обмена гликозаминогликанов, для которого характерен специфический внешний вид лица, аномалии глаз, костей, селезенки, печени. В некоторых случаях заболевание может сопровождаться задержкой умственного развития.
Существуют различные типы мукополисахаридоза, развитие каждого из которых обусловлено определенной генетической аномалией, которая оказывает влияние на синтез специфического фермента.
Существующие методы лечения мукополисахаридоза имеют низкий уровень эффективности.
Причины мукополисахаридоза
Причиной развития мукополисахаридоза является нарушение ферментативного катализа гликозаминогликанов в лизосомах. При мукополисахаридозе нарушается процесс расщепления и сохранения мукополисахаридов, которые являются основными компонентами соединительной ткани. Избыток мукополисахаридов проникает в кровь и накапливается в тканях. Поэтому данное заболевание относят к болезням накопления.
В настоящее время выявлено 10 генетических типов мукополисахаридоза, четыре из которых возникают при нарушении активности гликозидаз, пять – сульфатаз, один развивается в случае дефицита трансферазы.
Заболевание наследуется по аутосомно-рецессивному и рецессивному, сцепленному с Х-хромосомой типам.
Типы и симптомы мукополисахаридоза
Выделяют следующие типы мукополисахаридоза:
- I тип или синдром Гурлера;
- II тип или синдром Гунтера;
- III тип или синдром Санфилиппо;
- IV тип или синдром Моркио;
- V тип или синдром Шейе;
- VI тип или синдром Марото-Лами;
- VII тип или синдром Слая;
- VIII тип или синдром Ди Ферранте.
Симптомы мукополисахаридоза обнаруживаются у ребенка на первом году жизни и уже к 12-24 месяцам они становятся довольно выраженными.
У таких детей наблюдаются:
- Череп в форме лодочного киля;
- Шумное дыхание ртом;
- Грубые черты лица;
- Отставание в росте;
- Деформации скелета;
- Постепенное развитие сгибательных контрактур;
- Увеличение размеров живота;
- Пупочные и паховые грыжи;
- Гидроцеле;
- Изменения со стороны глаз: помутнение и увеличение размеров роговицы, врожденная глаукома, атрофия сосков зрительных нервов, застойные явления на глазном дне, пигментная дистрофия сетчатки;
- Снижение слуха;
- Изменения со стороны сердца;
- Умственная отсталость;
- Повышенный тонус мышц, нарушение координации движений, параличи.
При I типе заболевания симптомы мукополисахаридоза при рождении отсутствуют. Первые симптомы мукополисахаридоза в виде ограниченного разгибания пальцев на руках наблюдаются только в 3-6 лет. Со временем уменьшается объем движений в других суставах рук. К периоду полового созревания все симптомы проявляются особенно ярко.
II тип мукополисахаридоза чаще встречается у мальчиков. Для больных характерны: скафоцефалия, грубые черты лица, низкий грубый голос, шумное дыхание, частые ОРВИ. В 3-4 года возникают нарушения координации движений. Наблюдается прогрессирующая тугоухость, остеоартриты, узелковые поражения кожи спины.
При наличии синдрома Санфилиппо (III тип) ребенок в течение 3-5 лет нормально развивается, хотя иногда могут наблюдаться затрудненное глотание и неуклюжая походка. После трех лет у ребенка начинает развиваться апатия, возникает задержка психомоторного развития, нарушается речь, грубеют черты лица, возникает недержание кала и мочи, ребенок перестает узнавать окружающих. Такие дети умирают в возрасте 10-20 лет из-за интеркуррентных инфекций.
IV тип мукополисахаридоза характеризуется появлением первых симптомов в 1-3 года. Наблюдаются: резкая задержка роста, непропорциональное телосложение, грубые черты лица, кифоз или сколиоз, деформация грудной клетки; контрактуры в плечевых, локтевых, коленных суставах; плоскостопие; снижение мышечной силы; снижение слуха; дистрофия роговицы. Больные не доживают до 20-летнего возраста по причине сердечно-легочной недостаточности.
Для синдрома Шейе (V тип) характерен низкий рост, уплощенная переносица, короткая шея, контрактуры суставов, гипотония мышц конечностей, вегетативная лабильность, снижение сухожильных рефлексов, значительное помутнение роговиц.
Синдром Марото-Лами (VI тип) впервые проявляется после 2 лет. Он характеризуется отставанием в росте, грубыми чертами лица, бочкообразной грудной клеткой, малыми размерами верхней челюсти, короткой шеей, сгибательными контрактурами суставов рук. Интеллект больных не страдает.
Мукополисахаридоз VII типа устанавливают лишь при детальном биохимическом исследовании.
Синдром Ди Ферранте (VIII тип) схож с синдромом Моркио, но отличается выраженной задержкой интеллектуального и психомоторного развития.
Диагностика мукополисахаридоза
Диагностика мукополисахаридоза основывается на его характерных проявлениях, результатах рентгенологического исследования, установления экскреции гликозаминогликанов с мочой, исследования активности ферментов в фибробластах кожи.
Диагностировать мукополисахаридоз можно еще до рождения ребенка, используя для анализа амниотическую жидкость или ворсины хориона.
Лечение мукополисахаридоза
В настоящее время специфического лечения мукополисахаридоза не разработано.
Как правило, терапия мукополисахаридоза носит симптоматический характер. Пациента при этом наблюдают врачи различных специализаций: педиатр (по причине частых острых респираторных вирусных инфекций), хирург (в связи с наличием грыж), ортопед (в связи с нарушениями опорно-двигательного аппарата), отоларинголог (в связи с хроническими синуситами и отитами, нарушениями слуха), офтальмолог, невропатолог, нейрохирург.
В лечении мукополисахаридоза используются различные гормональные препараты (глюкокортикоиды, кортикотропин, тиреоидин), витамин А, цитостатики, сердечные препараты, переливания лейкоцитарной массы, крови, плазмы. Но все это вызывает лишь временные улучшения.
При синдроме Гурлера может проводиться трансплантация костного мозга. При ранних сроках ее проведения (до 1,5 лет) она может в значительной мере улучшить прогноз болезни, но данная процедура может вызывать ряд осложнений.
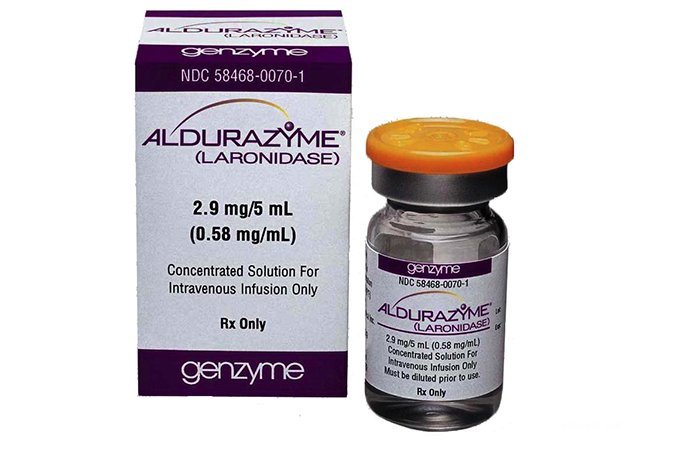
В США, странах Европы и Японии для лечения мягких форм мукополисахаридоза I типа применяется препарат альдуразим, который замещает недостающий в организме пациента фермент и приводит к улучшению состояния дыхательной системы, костей и суставов. Но внутривенное введение препарата не останавливает поражения ЦНС, поскольку альдуразим не проникает из крови в мозг. Данное средство применяется в случаях, когда трансплантация невозможна или непосредственно перед трансплантацией костного мозга, чтобы улучшить состояния больного и замедлить развитие болезни.
Таким образом, мукополисахаридоз – это редкое заболевание с неблагоприятным прогнозом для жизни пациента, поскольку с течением времени проявления болезни только усиливаются, а эффективного способа ее лечения пока нет. Единственный способ предотвратить заболевание – это обнаружить его еще в неонатальном периоде и предпринять соответствующие меры.