Синдромом Марфана называют патологию, характеризующуюся недоразвитием соединительной ткани у детей в эмбриональном или постнатальном периоде, что обусловлено структурными дефектами коллагена и сопровождается поражением преимущественно опорно-двигательного аппарата, сердечно-сосудистой системы и глаз.

Согласно данным разных авторов, частота встречаемости этого заболевания составляет 1 случай на каждые 10-20 тыс. новорожденных, без половой и расовой принадлежности.
Причины патологии
Синдром Марфана – это врожденная аномалия развития, наследуемая по аутосомно-доминантному типу. В основе патологии лежит мутация в гене FBN1, который отвечает за синтез фибриллина – структурного белка межклеточного матрикса, являющегося важнейшим соединением, придающим эластичность и сократимость соединительной ткани.
Таким образом, основной причиной развития синдрома Марфана является дефицит фибриллина, который приводит к нарушению формирования волокнистых структур, потере упругости и прочности соединительной ткани и, как следствие, к невозможности выдерживать физиологические нагрузки.
Примерно в 75% случаев обнаруживается семейный тип наследования этого синдрома, в остальных 25% – первичная мутация.
Согласно исследованиям, чем больше возраст отца (особенно после 35 лет), тем выше риск рождения ребенка с синдромом Марфана.
Классификация заболевания
Синдром Марфана подразделяют на несколько видов в зависимости от количества пораженных у человека систем на:
- Стертую форму, характеризующуюся слабовыраженными изменениями в одной-двух системах;
- Выраженную форму, которая характеризуется изменениями хотя бы в одной системе; патологиями в двух и более системах; слабовыраженными нарушениями в трех системах.
По характеру течения синдром подразделяется на стабильный и прогрессирующий.
Признаки синдрома Марфана
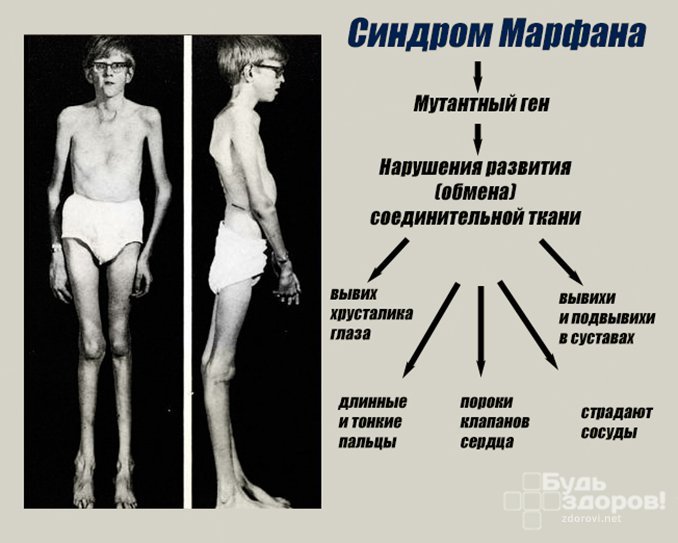
Клинической картине данной патологии свойственны поражения многих и самых разных жизненно важных органов и систем организма: скелета, глаз, нервной и сердечно-сосудистой систем.
В большинстве случаев признаками синдрома Марфана являются:
- Высокий рост;
- Относительно короткое туловище;
- Непропорционально длинные и тонкие конечности (долихостеномелия);
- Удлиненные паукообразные пальцы (арахнодактилия);
- Астеническое телосложение;
- Мышечная гипотония;
- Слаборазвитая подкожная клетчатка;
- Длинный и узкий лицевой скелет (долихоцефалия);
- Высокое аркообразное нёбо;
- Нарушение прикуса.
В среднем длина тела у девочек с синдромом Марфана составляет 52,5 см при рождении, окончательный рост – 175 см, у мальчиков – 53 и 191 см соответственно.
Также этому заболеванию свойственны нарушения функций суставов (гипермобильность), деформация позвоночника (кифоз, вывихи и подвывихи шейного отдела, сколиоз, спондилолистез, кифосколиоз), протрузия вертлужной впадины, плоскостопие.
В клинической картине синдрома доминирует сердечно-сосудистая патология, которая часто и определяет исход заболевания. Проявляется пороками развития перегородок и клапанного аппарата сердца, дефектами структуры стенок сосудов, в частности крупных ветвей легочной артерии и аорты, а также врожденными пороками сердца: стенозом легочной артерии, дефектом межпредсердной и межжелудочковой перегородки, коарктацией аорты и т.д.
Самая неблагоприятная форма синдрома Марфана, проявляющаяся уже при рождении в классическом варианте, приводит к прогрессирующей сердечной недостаточности и, как следствие, к летальному исходу в течение первого года жизни.
В большинстве случаев признаком синдрома Марфана является и патология органа зрения, характеризующаяся близорукостью, косоглазием, гипоплазией цилиарной мышцы и радужной оболочки, увеличением размера и уплощением роговицы, эктопией хрусталика, изменением калибра сосудов сетчатки.
Также у детей с этой патологией диагностируется поражение нервной и бронхолегочной систем и других органов: эктопия почек, разрывы и вывихи связок, варикозное расширение вен, бедренные и паховые грыжи, опущение мочевого пузыря и матки у девочек, поражение кожи и мягких тканей.
Синдрому Марфана характерен высокий выброс адреналина, что является причиной гиперактивности, нервного возбуждения, а иногда умственной одаренности и развития неординарных способностей.
Диагностика синдрома Марфана
Окончательный диагноз может быть поставлен только после комплексного обследования пациента, в котором принимают участие генетик, ортопед, кардиолог и офтальмолог.
Целью генетиков является изучение семейной истории и в том числе выявление близких родственников, которые умерли от сердечно-сосудистых заболеваний. С целью диагностики синдрома Марфана кардиолог назначает эхокардиограмму, ЭКГ, рентген грудной клетки. Офтальмолог обследует глаза с помощью щелевой лампы с целью выявления вывиха хрусталика или каких-либо других патологий. Ортопед обследует грудную клетку и позвоночник на наличие сколиоза, плоскостопия и т.п.
Если подтверждается версия наследственности, то для постановки окончательного диагноза требуется наличие характерных признаков синдрома Марфана как минимум в двух системах организма. В том случае если наследственности нет, то необходимо подтверждение поражения хотя бы трех систем организма.
Лечение синдрома Марфана
Определенных методов лечения синдрома Марфана, к сожалению, не существует. Главная цель терапии – устранение сопутствующих патологий и предупреждение осложнений.
Лечение прогрессирующего сколиоза проводится физиотерапевтическими процедурами и механическим укреплением скелета, в отдельных случаях проводится хирургическая коррекция.
Некоторым больным показана хирургическая пластика аорты, аортального и митрального клапанов.
Коррекция зрения осуществляется с помощью очков или линз, при необходимости назначается лечение катаракты, глаукомы и т.д.
Таким образом, всем пациентам показана симптоматическая терапия.