Синдром Прадера-Вилли является редкой наследственной болезнью, возникающей из-за отсутствия отцовской копии одного из участков хромосомы. Впервые он описан в середине ХХ века швейцарскими учеными А. Прадером и Х. Вилли. Заболевание встречается у 1:12000-15000 рожденных детей.
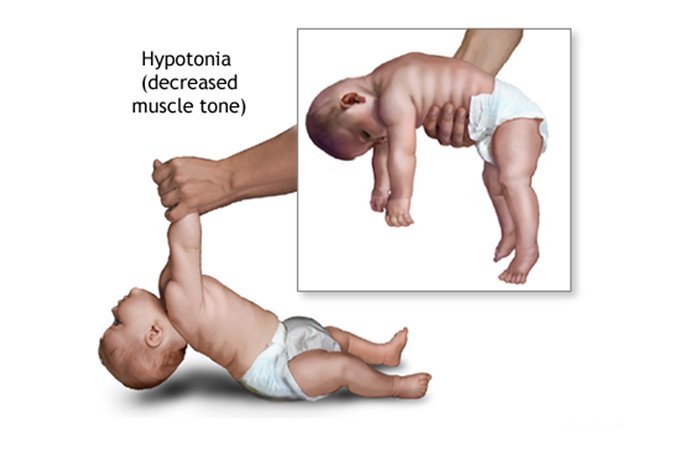
Клиническая характеристика синдрома Прадера-Вилли
Дети с этой наследственной болезнью обычно рождаются доношенными. Часто у них наблюдается нерезко выраженная внутриутробная гипотрофия (низкая подвижность плода). Врачи различают 2 фазы синдрома Прадера-Вилли:
- Первая характеризуется выраженной мышечной гипотонией. Часто ее дополняют: рефлекс Моро, снижение сухожильных рефлексов, тенденция к гипотермии;
- Вторая фаза болезни обычно развивается через несколько месяцев и проявляется в виде полифагии. Дети не могут утолить свой голод, поэтому они готовы есть непрерывно, что приводит к развитию ожирения. При этом жир откладывается чаще всего на проксимальных отделах конечностей и на туловище, в то время как кисти и стопы диспропорционально маленькие. Во второй фазе признаки гипотонии несколько уменьшаются.
У таких детей рост обычно снижен. У мальчиков возникает гипоплазия мошонки, полового члена, крипторхизм. Девочкам характерны гипоплазия половых губ, а в более взрослом возрасте –аменорея и гипоплазия матки.
Как правило, у больных детей психомоторное развитие отстает от нормы. У них хорошая зрительная долговременная память, и они могут научиться читать, но их собственная речь обычно отстает от понимания. Математические навыки, слуховая память и навыки письма чаще всего развиты хуже, что требует особого подхода при обучении. Синдром Прадера-Вилли у детей характеризуется затрудненной речью и маленькимсловарным запасом. Как правило, больные доброжелательны по характеру, плохо умеют управлять своими эмоциями, безынициативны, и им свойственны резкие перепады настроения.
Помимо этих основных симптомов может наблюдаться:
- Высокое арковидное небо;
- Сухая слизистая полости рта;
- Микроцефалия;
- Кариес и дефекты эмали;
- Гипоплазия хрящей ушных раковин;
- Судороги и страбизм;
- Микродонтия;
- Мезобрахифалангия;
- Сколиоз;
- Клинодактилия;
- Синдактилия;
- Нарушение координации;
- Поперечная ладонная складка.
Также нередко в период совершеннолетия синдрому сопутствует сахарный диабет, аномальная гибкость и жидкие лобковые волосы. Обычно у больных встречается не больше пяти указанных признаков.
Общими внешними признаками взрослых с синдромом Прадера-Вилли являются:
- Широкий и большой нос;
- Избыточный вес с жировыми отложениями в центральной части тела;
- Чувствительная кожа, на которой легко образуются синяки и царапины;
- Маленькие ноги и руки с непропорционально узкими пальцами.

Диагностика и лечение синдрома Прадера-Вилли
Диагностика синдрома Прадера-Вилли проводится на основании симптомов болезни, которые подтверждают генетическим анализом. Такое исследование рекомендуется проводить всем детям с пониженным мышечным тонусом.
Необходимо отметить, что при помощи обычного исследования хромосомного состава кариотипа диагностировать унипарентальную дисомию или микроделецию нельзя. Для этого применяют специальные молекулярно-генетические и цитогенетические методы – прометафазный анализ, а также используют ДНК-маркеры определенных участков хромосомы 15 и некоторые другие.
Сегодня синдромы Ангельмана и Прадера-Вилли приняты за общую модель для изучения новых и сложных явлений клинической генетики – унипарентальной дисомии и геномного импринтинга.
Без проведения генетических исследований может быть установлен неправильный диагноз –синдром Дауна или миопатия. Однако опытный генетик обычно точно определяет болезнь, поскольку такие дети, как правило, имеют много схожих признаков.
После проведения диагностики синдрома Прадера-Вилли врач назначит лечение, направленное на повышение качества жизни. Обычно лечение синдрома Прадера-Вилли включает массаж и другую специальную терапию. Также рекомендованы занятия с дефектологом и логопедом и использование различных методик развития ребенка. Довольно часто применяется медикаментозная терапия, включающая прием «гормонов роста» и проведение заместительной гормональной терапии с применением гонадотропинов.
На фоне неопущения яичек и микропении у мальчиков может потребоваться хирургическое вмешательство или гормонотерапия. Для того чтобы корректировать повышенный вес, следует соблюдать диету с ограничением количества углеводов и жиров.
Поскольку синдром Прадера-Вилли – генетическая болезнь, терапия может только улучшить качество жизни больного. И чем раньше будет поставлен диагноз, тем больше шансов сохранить здоровье и создать условия для социальной адаптации больных.